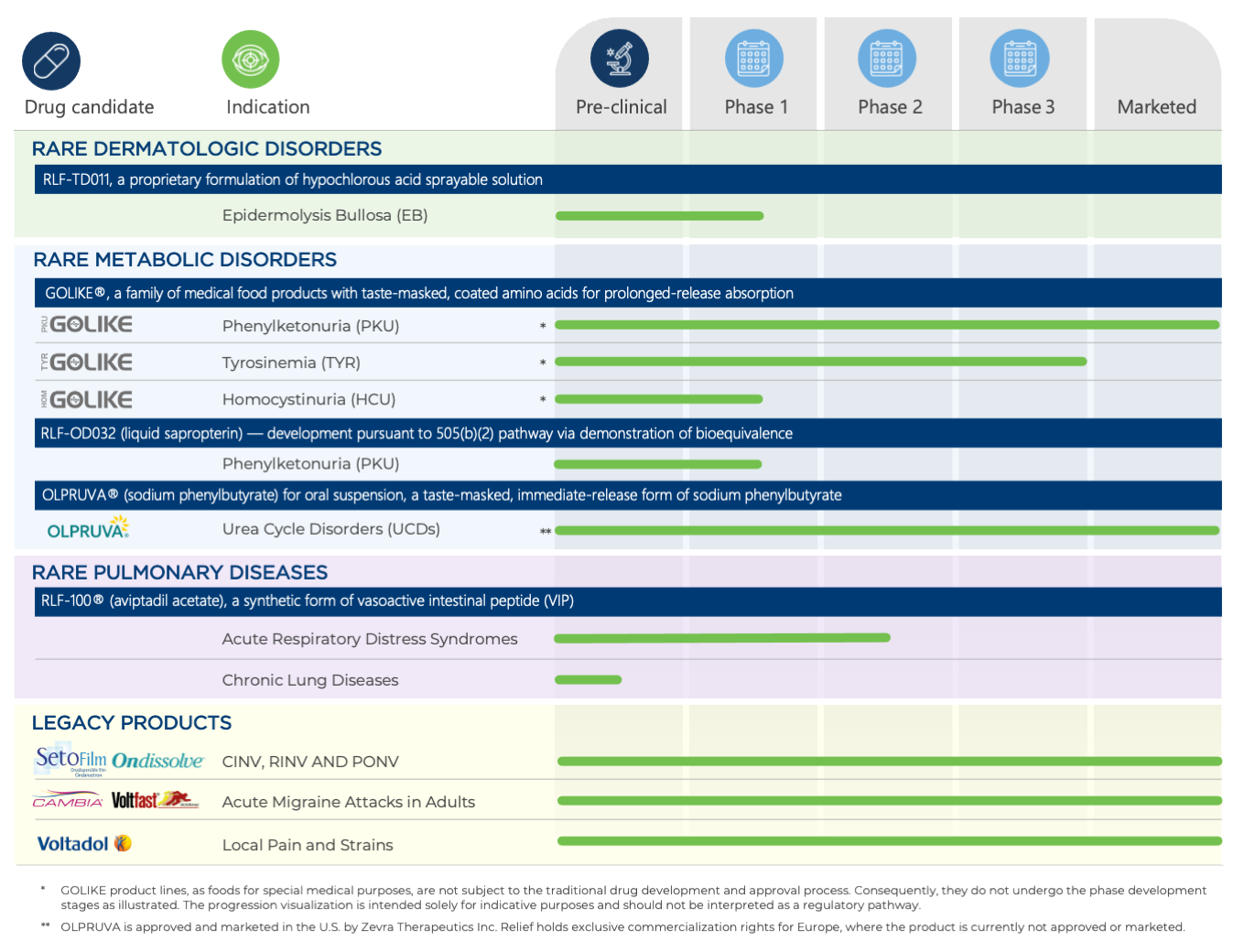
PIPELINE AND PORTFOLIO
We are dedicated to advancing transformative therapies to address unmet medical needs and improve the quality of life for patients living with chronic and debilitating conditions. Relief's diversified portfolio combines a pipeline of promising drug candidates with revenue-generating products marketed through established partnerships.
PIPELINE
RLF-TD011 for the Treatment of Epidermolysis Bullosa 
RLF-TD011 is in clinical development for the treatment of epidermolysis bullosa (EB), a group of rare genetic skin disorders that cause the skin to blister and tear with minimal friction. RLF-TD011 is a differentiated hypotonic, acid-oxidizing solution formulated with hypochlorous acid (HCIO), offering strong antimicrobial and anti-inflammatory properties. It has been granted Orphan Drug Designation (ODD) by the U.S. FDA for the treatment of EB, which qualifies the sponsor of the treatment for certain development incentives, including seven years of market exclusivity upon FDA marketing approval.
Relief is developing RLF-TD011 as a fast, easy-to-use, and effective EB wound care therapy designed to efficiently control infection and inflammation while reducing antibiotic use and easing the intensive wound care routine required by current treatments. Importantly, RLF-TD011 may also enhance the efficacy and usability of newly developed EB treatments.
RLF-OD032 for the Treatment of Phenylketonuria
Leveraging our expertise in the development and commercialization of treatments for metabolic disorders, we initiated the development of RLF-OD032 in 2022 as a next-generation treatment for phenylketonuria (PKU), a rare genetic metabolic disorder.
RLF-OD032 is an innovative, highly concentrated liquid formulation of sapropterin dihydrochloride for oral administration, designed to address the limitations of certain current treatments for PKU in adult and pediatric patients. We are pursuing U.S. regulatory approval under the 505(b)(2) NDA submission pathway, demonstrating bioequivalence to a reference listed drug. If approved, RLF-OD032 would be the first and only ready-to-use, portable, and liquid sapropterin drug, which may significantly improve PKU management by enhancing treatment compliance and quality of life.
RLF-100® for the Treatment of Pulmonary Indications
Aviptadil acetate is a synthetic form of vasoactive intestinal peptide, a naturally produced peptide with anti-inflammatory, immunosuppressive, anti-proliferative, and vasodilatory properties. Relief has developed proprietary, patent-protected Aviptadil formulations (RLF-100®) for intravenous and inhaled administration.
RLF-100® has the potential to address significant unmet medical needs in patients affected by acute respiratory distress syndromes (ARDSs) and certain chronic lung diseases, including sarcoidosis, berylliosis, and checkpoint inhibitor-induced pneumonitis. We are actively seeking partnerships or collaborations to support further development and potential commercialization of RLF-100®.
ACER-001 for the Treatment of UCDs
ACER-001 is a proprietary, taste-masked formulation of sodium phenylbutyrate powder, owned and developed by Acer Therapeutics, a wholly owned subsidiary of Zevra Therapeutics (NASDAQ: ZVRA). ACER-001 is approved and marketed by Zevra Therapeutics in the United States as OLPRUVA®, indicated as an adjunctive therapy for the long-term management of urea cycle disorders involving deficiencies in carbamylphosphate synthetase, ornithine transcarbamylase, or argininosuccinic acid synthetase.
Relief holds exclusive development and commercialization rights for ACER-001 in Europe, where it is currently not approved or available.
COMMERCIAL
PKU GOLIKE®
Relief developed PKU GOLIKE® , the first prolonged-release medical food for the dietary management of PKU. Characterized by a special coating that enables more physiological absorption, mirroring the absorption profile of natural proteins, PKU GOLIKE® offers enhanced metabolic control and palatability compared to standard amino acid protein substitutes.
In the United States, PKU GOLIKE® is exclusively marketed by Eton Pharmaceuticals (NASDAQ: ETON) under a licensing agreement with Relief. Since January 2025, intellectual property and commercialization rights for PKU GOLIKE® outside the U.S. have been held by Nutrisens. Relief remains the exclusive global supplier of PKU GOLIKE® and continues to develop product line extensions in PKU and other metabolic disorders.
Diclofenac Formulations
Relief has developed a diverse portfolio of diclofenac formulations for multiple pain and inflammatory conditions, including migraine, arthritis, and localized pain. Our out-licensed formulations are marketed by third parties under established brands, including CAMBIA® (Assertio Therapeutics), Voltfast® (Novartis), Diclo-ratiopharm® (Ratiopharm), Potafast® (DLBEEN), and Voltadol® (Fidia Farmaceutici).
SETOFILM®/ONDISSOLVE®
SETOFILM® and ONDISSOLVE® are an orodispersible film formulation of ondansetron, developed by Relief for the prevention and treatment of nausea and vomiting associated with radiotherapy-induced (RINV), chemotherapy (CINV), and postoperative sequels (PONV).
Relief out-licensed the rights in Canada to Takeda Pharmaceuticals, which markets the product as ONDISSOLVE® . Commercial and intellectual property rights in Europe, Australia and New Zealand, were acquired in 2020 by Norgine B.V., which markets the product as SETOFILM®.
For additional information on the Company's products, development programs, and targeted indications, please refer to the pipeline and portfolio section of Relief's latest Annual Report and the Company's most recent press releases.